1. Gas Sorption by Water Droplets
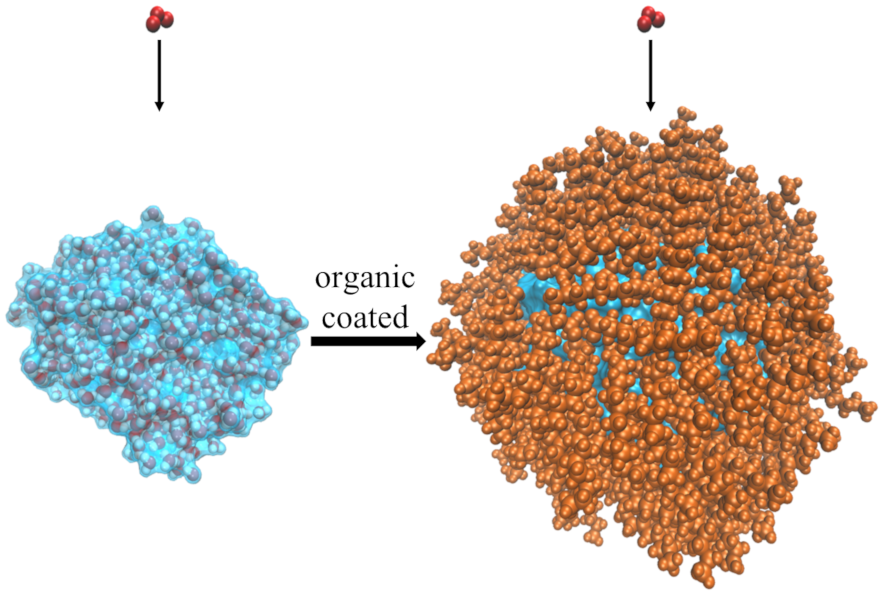
Gas sorption by atmospheric aerosols, such as water droplets, can significantly alter the radiation and chemical properties of the atmosphere. Such processes can be further complicated by the presence of organic species commonly found in the atmosphere. Our research in this project focuses on understanding the mechanism of gas sorption by pure/organic-coated water droplets on the molecular scales with the help of Molecular Dynamics (MD) simulations.
2. Water Droplet Coalescence
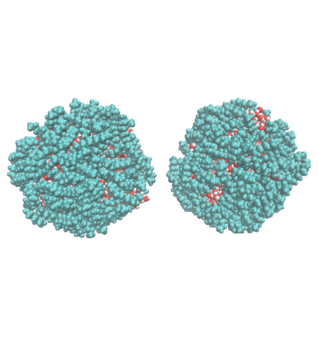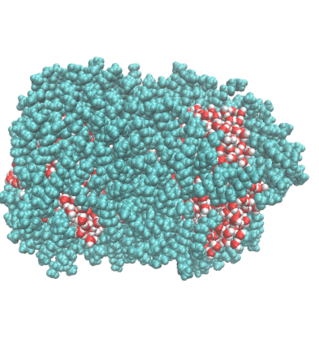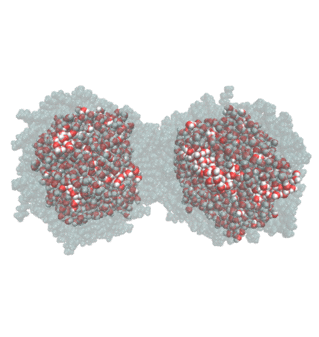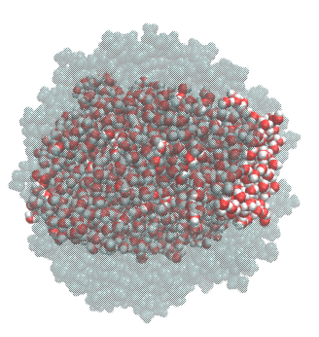
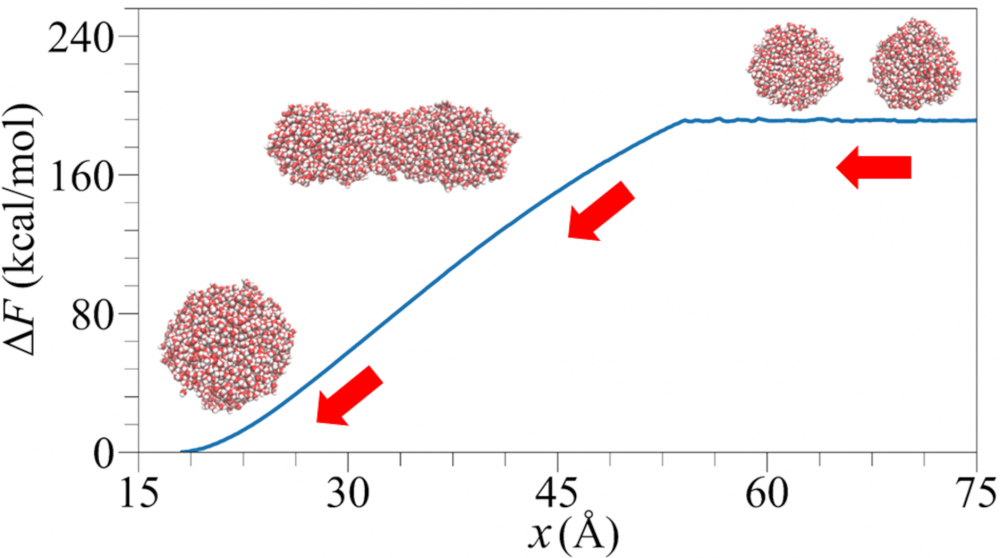
Droplet coalescence is a process in which two or more particles/droplets merge to form a larger one permanently. It has received great attention in recent years due to their importance in cloud droplet growth and raindrop formation in atmospheric sciences. In addition to the coalescence of pure water droplets, how organic matters in the atmosphere affect coalescence is also a major concern. In this project, we are interested in understanding coalescence by understanding the thermodynamics as well as the dynamics via molecular simulations.
3. Patchy colloidal particle at the fluid-fluid interfaces
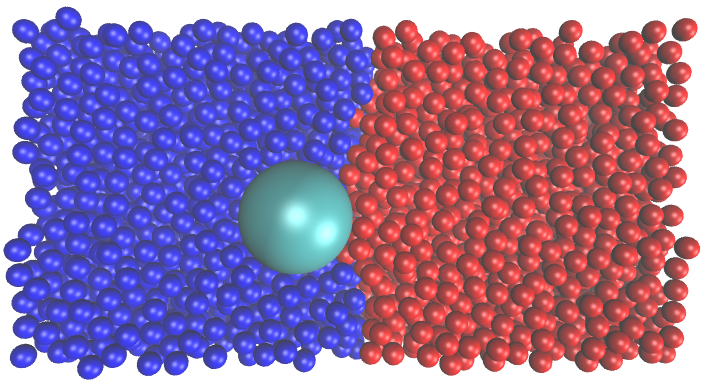
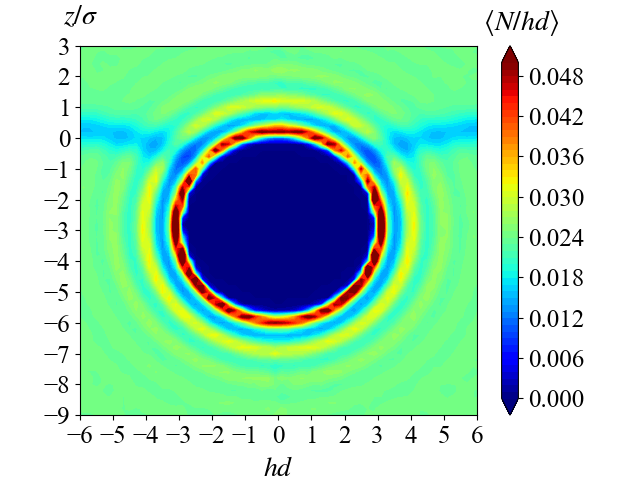
Colloidal particle adsorption at fluid-fluid interfaces has been long known to have emulsion stabilizing effects. The studies of the fluid-fluid interfaces have gained traction thanks to its wide variety of industrial applications, such as food production, electronics, textiles, oil refinement, and catalysis. The fluid-fluid interface can also be used as a template for building nanostructures that is important for the development in nanotechnology. Recent advances in synthetic methods have allowed productions of patchy particles with different geometry and surface anisotropy that can self-assemble into a wide variety of interesting nanostructures. To contribute to the field of colloidal particle research, we study the stability and dynamics of different colloidal particle systems at the fluid-fluid interface as well as the fluid-solid interface by both Monte Carlo (MC) and Molecular Dynamics (MD) simulations.
4. Chemical kinetics of catalyzed asymmetric organic reactions (in collaboration with Prof. Yeung-Ying YEUNG)
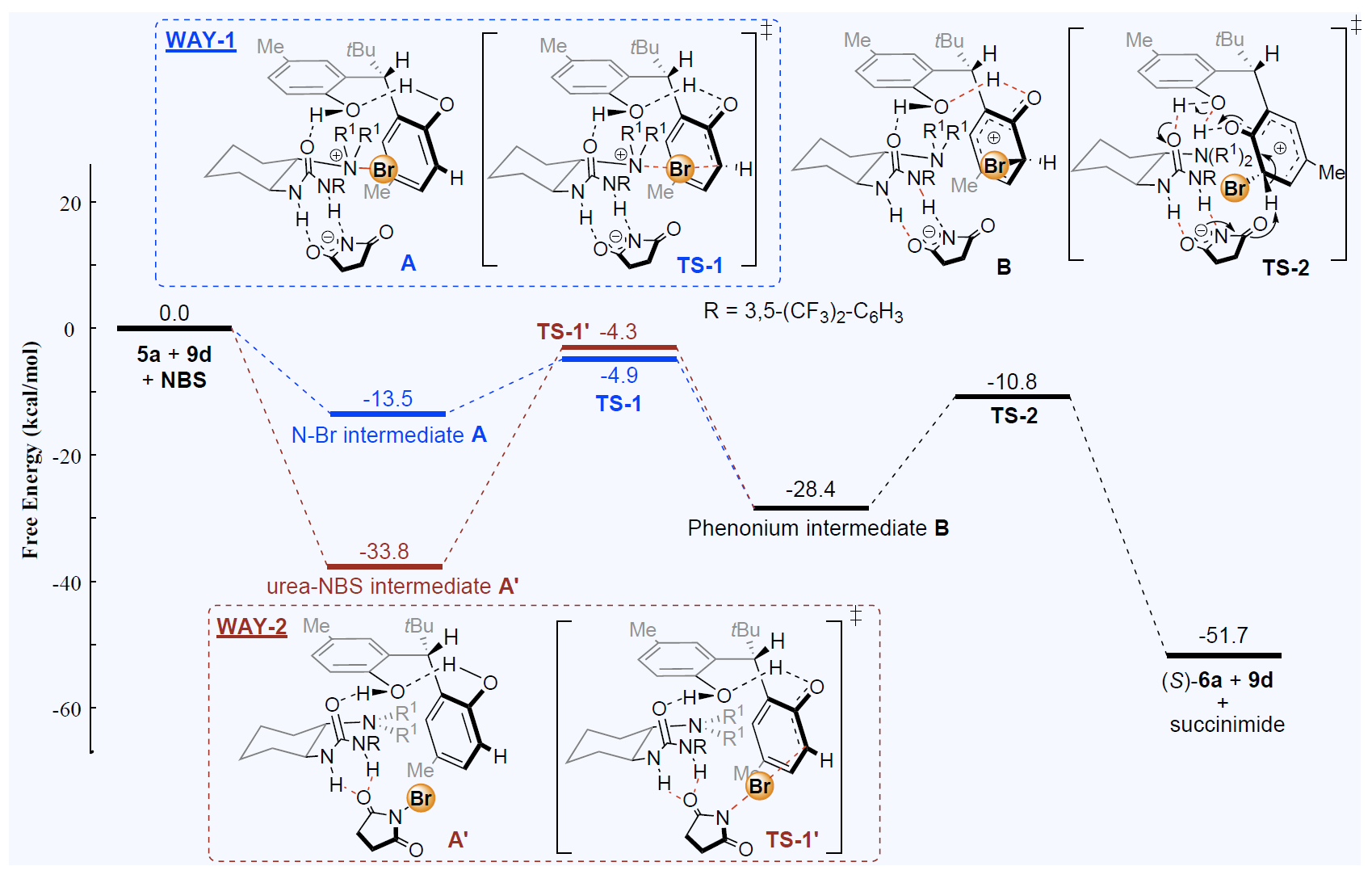
With the help of theory and computation, we can further our chemical insight into catalysts and improve them. In collaboration with Prof. Yeung-Ying YEUNG at CUHK, our group uses a variety of quantum chemistry tools including electronic structure calculations and ab initio molecular dynamics (AIMD) to understand the reaction mechanisms of how different organic catalysts carry out their functions in asymmetric synthesis, in which specific chiral compounds are made and separated. Our group is also interested in applying multi-scale modeling, using theories based on statistical mechanics, to account for explicit solvent effects.
Ying-Lung Steve Tse
Room 246 Science Centre
Office: (852) 3943 6373
stevetse@cuhk.edu.hk